苏龙菊同学搭建合理模型发表姊妹篇,预测高熵合金催化剂在析氧反应和析氢催化活性
姊篇:HER
A comprehensive theoretical study on hydrogen evolution reaction mechanism of noble metal-based multi-principal element alloys with broad pH applicability based on atomic site preference behavior
基于组成金属原子在亚晶格上的占位偏好以及中间体吸附位点偏好
来预测FCC结构多主元合金FCC_CoCuFeNiPd and CoCuFeNiRu在宽域PH值环境下的析氢反应催化活性
Longju Su, Minliang Gao, Liangji Weng, Cheng Qian, Xiaoqiong Zhang, Xingyu Chen, Rong Chen, Jiansen Wen, Bo Wu, A comprehensive theoretical study on hydrogen evolution reaction mechanism of noble metal-based multi-principal element alloys with broad pH applicability based on atomic site preference behavior, International Journal of Hydrogen Energy, Volume 234, 15 May 2026, 155167, https://doi.org/10.1016/j.ijhydene.2026.155167
Abstract
Noble metal-contained multi-principal element alloys (MPEAs) are potentially and widely used in various catalytic applications due to their excellent catalytic performance. This study predicted the atomic site preference and hydrogen evolution reaction (HER) performance of CoCuFeNiPd and CoCuFeNiRu alloys with face-centered cubic (FCC) structure. Catalytic activity was analyzed based on atomic site occupying fractions (SOFs), with special quasi-random structures (SQS) generated for comparison. Hydrogen adsorption free energies 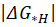 of Slab models with SOFs and SQS were calculated to evaluate catalytic performance. The SOFs-structured CoCuFeNiPd showed superior catalytic activity with an ultra-low of 0.006 eV, outperforming CoCuFeNiRu (0.027 eV). Both alloys exhibited efficient water dissociation under alkaline/neutral conditions with energy barriers (0.52 eV and 0.84 eV) lower than that of Pt (0.95 eV), highlighting their excellent catalytic performance. In summary, the CoCuFeNiPd and CoCuFeNiRu MPEAs exhibited exceptional HER catalytic activity across a broad pH range.
of Slab models with SOFs and SQS were calculated to evaluate catalytic performance. The SOFs-structured CoCuFeNiPd showed superior catalytic activity with an ultra-low of 0.006 eV, outperforming CoCuFeNiRu (0.027 eV). Both alloys exhibited efficient water dissociation under alkaline/neutral conditions with energy barriers (0.52 eV and 0.84 eV) lower than that of Pt (0.95 eV), highlighting their excellent catalytic performance. In summary, the CoCuFeNiPd and CoCuFeNiRu MPEAs exhibited exceptional HER catalytic activity across a broad pH range.
妹篇:OER
基于组成金属原子在亚晶格上的占位偏好以及中间体吸附位点偏好
来预测FCC结构多主元合金FCC_CoCuFeNiPd and CoCuFeNiRu的析氧反应催化活性
该研究结果进一步说明了多主元合金原子分布和中间体吸附位点模型的合理搭建的必要性、重要性和可行性。否则不合理模型,将导致预测结果偏差太大,而误导新材料研究开发。综合评价还得进行更全面计算,因为实验上不知暴露出哪些晶面指数,肯定不都是最想要的那个面。与别的吃大锅饭,随机分布模型相比,我们的工作应该是说更深刻和合理,但本文也没做太全面和完善,主要是全面展开计算,任务太重,烧不起,可略作参考。
Longju Su, Hehua Que, Cheng Qian, Xiaoqiong Zhang, xingyu chen, Rong Chen, Jiansen Wen and Bo Wu, Prediction of oxygen evolution reaction activity of FCC_CoCuFeNiPd and CoCuFeNiRu multi-principal element alloys based on sublattice preference of constituent atoms and the site preference of intermediates, Physical Chemistry Chemical Physics, DOI:10.1039/d5cp02461g
Abstract:Noble metal-contained multi-principal element alloys (MPEAs), primarily composed of less noble elements, emerge as promising catalysts by enhancing catalytic performance while reducing cost compared with conventional noble metal catalysts. However, limited understanding of the underlying catalytic mechanisms has hindered the exploration of practical applications of MPEA catalysts due to the complex atomic environments, unique surface structures, and synergistic effects associated with MPEAs. In this work, the oxygen evolution reaction (OER) performance of FCC_CoCuFeNiPd and FCC_CoCuFeNiRu was theoretically investigated in detail under alkaline conditions, based on the sublattice preference of constituent atoms and the site preference of intermediates. For this purpose, firstly, the site occupying fractions (SOFs) of the constituent elements on the sublattices were predicted by combining the sublattice model with computational thermodynamics. Then, the reaction surfaces were cleaved from the bulk alloy models, which allowed a richer set of adsorption and reaction sites to be systematically considered. This approach contrasted with the commonly used, but oversimplified, special quasi-random structure (SQS) model, in which all constituent alloying elements were assumed to be perfectly randomly distributed on different sublattices, i.e., the vertex positions (1a) and face-centered positions (3c) of the ordered cubic crystal lattice. Based on the advanced model, we demonstrated that CoCuFeNiRu outperformed CoCuFeNiPd, exhibiting a lower average overpotential (0.210 V vs. 0.325 V) and a more favorable optimal surface overpotential (0.181 V vs. 0.214 V). This was determined by calculating the Gibbs free energies of *OH, *O, and *OOH intermediates individually at 91 adsorption sites on the Slab surface. Ru exhibited adsorption free energies of intermediates closer to ideal values, facilitating reaction steps, whereas Pd’s higher free energies raised the overall overpotential. Meanwhile, we found that the average overpotential of CoCuFeNiPd based on the SOFs model (0.214 V) aligned better with the experimental value (0.193 V) than that predicted by the SQS model (0.25 V), highlighting the necessity and importance of considering the sublattice preferences of constituent alloying elements. Our contribution offers a theoretical foundation to explore and design high-performance noble metal-contained MPEA catalysts.